Multidimensional data often has a latent sequential structure that determines the basic shape of the data. Those sequential structure might correlate with ordered quantities like time, stages and size etc. Capturing and visualizing those sequential structure would be very helpful for exploratory data analysis.
A frequently used method for exploring high dimensional data is the principal component analysis (PCA). By projecting data to the main principal components, we can gain institutive information about the high level large scale shape of the data. More particularly, the projection to the first principal component offers a way to visualize the main back-bone linear structure.
A main short-coming of PCA is that it only structure from linear perspective. It fails for data whose latent sequential structure significantly deviate from linearity. For instance, let's consider the 2-dimensional data set depicted by the following map:
This dataset consists of multiple clusters each possesses, more or less, a sequential structure. The PCA wouldn't be able to aggerate these partial sequential structure to a global sequential structure.
The t-SNE algorithm with the high fidelity modification offers a simple yet effective way to discover global sequential structure in high dimension data: we simplify apply t-SNE to reduce a given dataset to 1-dimensional space. The coordinates of data points in the 1-dimensional space then provide the keys to order data points. The following video clip illustrates how t-SNE works on the dataset mentioned above:
We notice in above video that t-SNE discovered, more or less, the sequential structure underlying the sub-clusters; and furthermore, those partial sequential structures are aligned/ordered in a coherent way.
Dual Sorting of Heatmaps
Heatmap is a popular method to visualize matrix of numerical data. Heatmap is often used together with dendrograms, which rearrange the rows and columns using certain hierarchic clustering algorithm. The following picture shows a typical heatmap together with two dendrograms:
Heatmap augmented with dendrograms (wikipedia)
Heatmap constructed with dendrograms offers intuitive visualization, but it has difficulty to scale up to large dataset. For large matrix like those from single cell RNA sequencing (scRNA seq) study, dendrogram become quite inefficient, and instable in the sense that small change in data might lead to large variation in the clustering structure.
The t-SNE based sorting algorithm described in this note provides a simple alternative to arrange rows and columns in a heatmap: we simply sort the rows and columns with t-SNE. As an example, I took a dataset from scRNA Seq analysis, it comprises the gene expression counts of about 11000 cells with respect to 28000 genes. The following pictures shows its t-SNE embedding and its heatmap:
In above heatmap, each row represents the expression profile of a cell with respect to all selected genes; and each column represents the expression profile of a gene with respect to selected cells. Dark or red elements in the heatmap represent highly expressed cell/gene pairs. The t-SNE map is a embedding of all cells. We can see that several cell clusters are clearly visible as dark horizontal strips. However, no clear gene clusters are visible in the heatmap.
The following picture shows the heatmap after sorted its rows and columns with t-SNE. We see much more clearly the clusters among cells and genes, and more importantly the association between the cell- and gene-clusters.
It should be noticed with regards to the experiment above:
The perplexity of the t-SNE is set to about 2500. Smaller perplexity often leads to instability and random artifacts.
The training process with t-SNE has ran for 5000 epochs, which is much higher than normal t-SNE training process. Training less epochs likely results in more random artifacts.
In above example, t-SNE used the linear correlation as distance metric instead of the normally used Euclidean distance. Various experiments indicate that correlation seems to be more appropriate to measure similarity between genes. Because of this, we can not use PCA to pre-process data table to much lower dimensions to speed up the training, since PCA in general doesn't preserve correlation table.
Unfolding of t-SNE Embedding
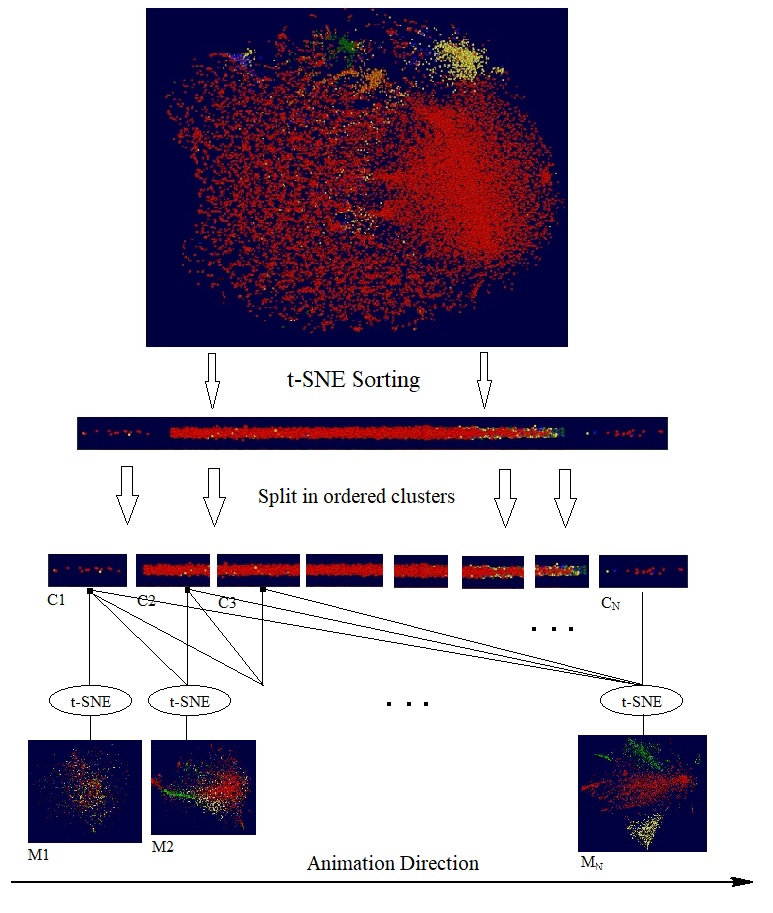
We have previously blogged about doing "knockout" test with t-SNE. By comparing the t-SNE maps produced by the different set of genes, we can explore the impact of disabled genes. Going one step further, with t-SNE as a multidimensional sorting algorithm we can perform the following experiment: We first sort all the participated genes into a sequence; then pick a particular direction for the sequence and split the sequence in small clusters; then create t-SNE cells maps with progressively larger gene clusters. The following diagram illustrates these steps:
It should be noticed that the sequence of t-SNE maps are generated independently by separate training processes, and each of them started from a different random initialization. In other words, the final maps M1, M2, M3 are independent embeddings of ordered gene clusters, C1, {C1, C2}, {C1, C2, C3}, etc.. If we are lucky, the order underlying the gene clusters might indicate certain latent variables, like time. The following short video clip shows the animation of through the resulting t-SNE maps for the dataset in previous section.
It is interesting to notice that the animation is reminiscent of group of cells unfolding, generation by generation, to final shape and structure.
High Fidelity t-SNE with Gradual Diminishing Exaggeration
When we repeat t-SNE on the same dataset with the same parameters, we may get different maps with noticeable random variations. This kind of randomness limits the accuracy of t-SNE algorithm as a visualization tool.
In this note we describe a simple yet effective method to improve the accuracy of t-SNE: Instead of switching the exaggeration rate at a fixed time during the training, we propose starting the training process with a high exaggeration, say 12.0, then gradually reduce it to 1.0. We will demonstrate the effectively of this method with example from single cell RNA sequential study.
Post-Training Normalization
Since the cost function optimized by t-SNE is invariant under rigged transformations (i.e. rotations and shifting), t-SNE normally produced maps with random rotations and shift. These kind of randomness can be easily removed by post-training normalization. For this study, we propose the following steps to normalize t-SNE maps as illustrated in the following diagram with 4 sample t-SNE maps:
As shown in above diagram, a t-SNE map is normalized in 3 steps:
Centralizing: centralizing the map so that each of the 2D or 3D coordinate has zero-mean.
PCA based Alignment: Rotating the map, so that the x-axis aligns with the first eigen vector of the map; y-axis with the second eigen vector, and potentially the z-axis with the third eigen vector.
Moment based Flipping: Calculate the 0.5-th moment of the map along the x-axis according the following forms:
If the moment is negative, flip the map along the x-axis by multiple all x components with -1.0; Do the same with y-axis and potentially z-axis.
After normalization we can then measure the discrepancy between two t-SNE maps by summing up the Euclidean distances between the data points in both maps. For more convenience, we can just count number of the data points whose images points have distance from each other larger than certain values, say 40 pixels.
It should be pointed out that there are cases that the suggested method might fail. For instance, when the t-SNP maps are rotational symmetrical where no clear orientation is present, this method will have difficulty find common orientations. In the practice, we often encounter this case, when the perplexity parameter of t-SNE is too small, so that the resulting t-SNE map resemble a disc or a ball. Fortunately, this kind of issue can be easily fixed by increasing the perplexity.
Finally, it is interesting to notice that there is no need to scale the maps, since t-SNE with fixed parameter settings normally produces maps with roughly the same size.
Training with Gradual Diminishing Exaggeration
In order to improve the training speed and efficiency, t-SNE algorithm initially proposed to start the training with a high exaggeration factor, train the map for certain number of epochs; then switch the exaggeration factor to 1.0 to practically terminate the exaggeration phase. The following diagram shows the cost minimized during a typical t-SNE training process, the diagram has been overplayed with the exaggeration factor.
We see that the objection cost has been reduced rapidly for a short period after the exaggeration phased has ended, but then stayed basically unchanged during the rest of the training process. This behavior suggests that real learning only takes place when exaggeration factor changes. We propose thus the following function to gradually decrease the exaggeration factor:
In above formula, f(n) is the exaggeration factor at n-th training epoch; C is the initial exaggeration factor; N is the number of total training epochs; L is the length of exaggeration phase which is set normally to 0.9N.
Example
As example I picked a dataset from single cell RNA sequence (scRNA seq) study. The dataset contains the expression counts of about 22000 cells with respect to 28000 genes. The input data for the t-SNE is thus a 22000x28000 matrix. The following picture shows a heatmap and a t-SNE map of this dataset:
For the test, t-SNE has been run on the dataset 4 times with and without enabling the new exaggeration method. The following pictures shows the cost reduction during training of the four repeats:
We see that, with the new method enabled, the final t-SNE maps have consistently lower cost. We also notice that, with the new method, the final maps have almost equal costs, whereas with old method, the cost of final maps vary slightly. Those small variations are more visible in the final maps. The following two pictures show the maps created with and without the new method:
With some effort we can discern relative large variation amount the maps created with old exaggeration strategy. We can visualize those variations more clearly with animation by interpolation between those maps as be shown in the following video clip:
Knockout Test with High Fidelity t-SNE
Knockout test has been used in microbiology to probe gene functions during the development of organisms. For doing that, certain genes are disabled or knocked-out; and changes induced in the organism give indications about those knocked-out genes. In general, knockout test help us to trace the correlation between genotypes and phenotypes.
High fidelity t-SNE enables us to probe the effect of individual features on the output t-SNE maps. If consider the t-SNE maps as phenotypes derived from genotypes (i.e. gene expressions), we can perform kind of "knockout" test with t-SNE: We create first a t-SNE map with all features; then create a t-SNE map without certain features. We can then expect that the knocked out features would be responsible for the variation in the t-SNE maps.
The following short video demonstrates how "knockout" test might been done to study correlation between gene- and cell-clusters:
Conclusion
With a small change in its algorithm, t-SNE achieved much high accuracy and opens therefore doors for new applications.
Single-cell RNA sequencing (scRNA-seq) is a powerful tool to profile cells under given condition with respect to selected genes. As a contingency matrix, a scRNA-seq dataset records the the expression level of individual cells with respect to a set of genes or related transcriptical signatures. A widely used method to study such scRNA-seq data is embedding the expression profiles of cells, i.e. rows of such expression matrix, into low dimensional space, and visualizing them as 2D or 3D maps. Those maps offers an intuitive way to trace the phenotype clusters among cells. Yet, this kind of maps do not show the which cells are regulated by which genes, despite that the expression matrix is direct information about reglating relationship between cells and genes.
This note puts forward a method to embed expression profiles of cells and genes, ie rows and columns of the expression matrix, simultaneously into a single low dimensional space. The resulting map visualizes both cells and genes; and more interestingly, also the association between the clusters. The following diagram illustrates the relationship between the dual embedding and conventional embedding:
The dual embedding method uses the tSNE algorithm for the embedding. In particular, let C and G denote the set of cells and genes; dc and dg be distance functions over C and G respectively, With tSNE we can then obtain a cell map and a gene map from (C, dc) and (G, dg) respective as indicated in above diagram. As an extension to these methods, the dual embedding method embeds both C and G in to low dimensional space with tSNE. In order to do so, we define a distance function dcg over C⋃G as following:
In above formula, M is the expression matrix of the scRNA-seq dataset; M[x,y] stands for the expression level of x-th cell w.r.t. y-th gene; Mmax is the maximal element of M;h is a positive bias factor that additionally adjusts the distances between cells and genes.The last two terms in above formula define the distance between C and G in the sense that higher expression level between cells and genes will leads two lower distance. Additionally, larger bias factor h leads to larger separation between cells and gens.
As an example, I used a dataset with 12000 cells and 8000 genes from the data used in the publication tasic2018. The follow image shows a cell-genemap of this dataset. The expression matrix has been log-transformed; the perplexity for the tSNE algorithm is about 1000.0.
As a feature of embedding maps, we can expect that two closely located genes/cells have similar expression profile. Additionally, a gene (red dot) should have high expression level on closely located cells (green dots). As the cells and genes overlap each in the map significantly, the following image uses gif animation to blend-in the two groups successively into foreground.
Figure 1: Dual Embedding of cells and genes with tSNE Algorithm.
We notice here that the cells show a clear cluster structure, whereas the genes are rather less structured, except that a large gene clusters located on the far right side, those genes might be less relevant for the formation of these cells.
We also notice that, as shown in the following image, there are 4 or 5 cell clusters on the upper right corner whose nearby genes form a kind of sequential path. Those genes might play a special role in regulating those target cell clusters.
Embedding with Correlation Affinity
Notice that scRNA-seq datasets are basically contingency table of counts for cell/gene co-expression events, thus the nature of an expression matrix is more probabilistic, than spatial. For this reason, we purpose here a correlation distance for the embedding algorithm to replace the Euclidean distance.
More particularly, let M' be the row-normalized version of M as defined by
M'[x,y] := (M[x,y] - M[x, *])/||M[x,*]||
where M[x, *] and ||M[x,*]|| is the mean value and the norm of the x-th row of M respectively. Similarly, let M" be the column-normalized version M as defined by:
M"[x,y] := (M[x,y] - M[*,y])/||M[*,y]||
After the normalization, the correlation coefficient between two rows or two columns of M is just the scalar product the corresponding rows or columns in M' or M''. We now define our correlation distance pcg over C⋃G as following:
The operator ⦁ in above formula denotes the scalar product of two row or column vectors; h is as before a positive bias factor that additionally adjusts the distances between cells and genes. The default value for h is 1.0. Notice also that matrix M doesn't need to be log-transformed like the case for Euclidean distance function as the normalization scale the numbers in to common ranges.
As an example, the following picture shows the dual embedding of the data used in previous example by applying tSNE with the correlation distance pcg:
Figure 2: Dual Embedding of cells and genes with tSNE and correlation distance pcg.
Comparing to Figure 1, we notice that in this dual embedding, the genes show more clear and simple cluster structure; and there are much less overlapping between the clusters, albeit that quite some clusters intertwine with each other. In general, a dual embedding map has the property that genes are highly expressed in by nearby cells. As be shown in Figure 2, we can see that the most high density gene clusters are rather distant from the major cell clusters, this indicates that the majority of those genes probably don't participate in the cell regulation.
Dual Embedding with Eigengenes
Mathematically, eigengenes are the top principal components of the expression matrix with the largest eigen-values. Eigengenes are normally obtained with the SVD or PCA algorithm, they are widely used as a way to reduce the dimensionality of a dataset, and therefore reduce the complexity of calculations.
We can apply the dual embedding method to eigengenes to visualize their relationship with the cell clusters. The following picture shows a dual embedding of the cells of above example together with their 50 eigengenes, represented in purple color. We notice that the cells in this map form a similar cluster structure as that in Figure 2; whereas the genes are replaced by eigengenes in the neighborhood of the cell clusters. We can clearly see that most cell clusters are regulated by 2 or 3 nearby eigengenes.
This visual characteristics indirectly validate the correlation based distance as a proper choice for the dual embedding algorithm.
Conclusion
We have introduced a correlation based distance function for cell and gene expression profiles. With help of an embedding algorithm, like tSNE, we can obtain low dimensional map for both cells and genes. These dual embedding maps, not only shows the cluster among cells and genes, but also their correlation through their proximity in the maps.
As demonstrated with real examples, correlation in the sense of statistics seems to be a better metric to visualize events of different nature like cells and genes in the scRNA seq dataset; and in general large contingency table of counts. The dual embedding provides intuitive way to visualize and explore those data.
All simulations and examples in this note is done with the software package VisuMap and a custom plug-in module named Single Cell Analysis, which implements in particular the two distance functions described in this note. The plug-in can be downloaded and installed from VisuMap online.
Representation Learning for Algebraic Relationships
This note puts forward a simple framework to learn data representation with deep neural networks. The framework is inspired by mathematical representation theory where we look for vector representations for elements of algebraic structure like groups.
To describe the framework, let's assume that we have the following table for an abstract operation:
Table 1: Binary operation table for abstract items a, b, c and d.
Above table shows that the abstract entities a, b, c and d fulfill an abstract operation ⊕, for instance: a⊕a = 0; b⊕c = 3; etc.. We now try to find representations for a, b, c and d with two dimensional vectors. In order to do so, we start with a default zero-representation as in following table:
a:
(0, 0)
b:
(0, 0)
c:
(0, 0)
d:
(0, 0)
Table 2: Initial representation table for a, b, c, d as 2 dimensional zero vectors.
That means a, b, c and d are all represented by vectors (0, 0). We then construct a feed-forwards neural networks as illustrated as following:
The network has four input nodes to accept a pair of 2-dimensional vectors; and one output node to approximate the operation ⊕. Like conventional feed-forward neural network, the input and output nodes will get data from the operation and representation table before each optimization step. Unlike the conventional feed-forward network, the input nodes are configured as trainable variables which will be adjusted during the learning process; and their values will be saved back to the representation table after each learning step. More particularly, the learning process goes as follows:
Randomly select a sample from the operation table, say b⊕c = 3.
Lookup the representation for a and b from the representation table. Assign them to input nodes of the network. Lookup the expected output for b⊕c from the operation table, and pass it to the output node as learning target.
Run the optimization algorithm, e.g. the ADAM optimization algorithm, for one step, which will change the state of input nodes to new values b'0, b'1, c'0 and c'1.
Save b'0, b'1, c'0 and c'1 back to the representation table as new representation for b and c.
Continue with step 1.
With an appropriate choices for the network and training algorithm, the above training process will eventually converge to a representation for a,b,c,d, so that the network realizes the abstract operation as described in table 1. A tensorflow based implementation of above training process is available at Z4GroupLearning, which can be run on the google cloud computing platform from a browser.
Representation Learning for Transformations
In previous example the table 1 actually describes the finite group ℤ4, representation are linked to the input layer. In general, the framework is not restricted to binary operations and the representation vectors can be linked to any state vectors, i.e. weights, bias and other trainable variables, of the network.
For the demonstration we describe here a network that finds representation for transformations in the 3D space. Let's assume that X is a 1000x3 table consisting of 1000 points from a sphere in the 3-dimensional space. Let {T0, T1, ...} be a sequences of 3D-to-3D transformations which transform X to {Y0, Y1, ...}. Assuming that X and {Y0, Y1, ...} are known, we can then find representations for the transformations {T0, T1, ...} with a deep neural network as illustrated as following:
The transformations are linked to some internal part of the weight matrix. Before starting the training process, the transformations are initialized with zero-matrices with the same shape as the linked weight matrix. At each training epoch, a 3-tuple (X, Tt, Yt) is randomly selected; Tt is pushed to the linked weight matrix; then the optimization process is run to learn the map X-to-Yt mapping; After the optimization process has run for several loops for all data points in X and Yt, the linked weight matrix is pulled back in to the representation Tt, stored as T't for future epochs. The training process then continues with other randomly selected 3-tuples, until all Yt have sufficiently approximated by the network.
As an example, we have selected 30 linear translations and 30 rotations to transform the sphere in the 3D space. The following video clip shows the transformed images Yt:
After the training process has completed after about 100 epochs, we have got 60 matrices as representation for the transformations. We have embedded the 60 matrices into the 3-dimensional space with PCA (principal component analysis) algorithm. The following picture shows the embedding of the 60 transformations:
In above picture, each dot represents a transformation: a red dot corresponds to a linear translation, a yellow dot to rotation. We see that the representations generated by the deep neural network correspond very well the geometry structure of the transformations.
Representation Learning for Dimensionality Reduction Representation learning (RL) described here can be used to perform non-linear dimensionality reduction in a straightforward way. So for example, in order to reduce a m dimensional dataset to k dimension, we can simply take a feedforward network with k input nodes and m output nodes. As depicted in the following diagram, the input nodes are configured as learning nodes whose state values get values from the training data and adjusted during the learning process.
We notice that autoendcoder (AE) networks has often be used in various ways for dimensionality reduction and unsupervised learning. Comparing to AE, RL basically performs just decoding service. However, the essential difference is that the AE trains a network, whereas RL trains directly the lower dimentional representation.
The following maps shows an example of mapping a 3-dimenional sphere dataset to the 2 dimensional plane. An implementation with tensorflow on colab platform can be found here.
We notice that 2-dimenional map preserved large part of the local topology, while unfolding and flattening the 3D-sphere structure.
If encoding service (like that in AE) is needed, we can extend the above RL network with an encoding network that maps the network output to the lower dimensional data. This encoding network can be then trained independently from the decoding part, or concurrently with the decoding network part.
Application for Data Imputation in Epigenomics Epigenomic study shows that different cell types of multi-cellular organisms developed from a common genome by selectively express different genes. Many types of assays have been developed to probe those epi-genomic manipulations. The result of such an assay is normally a sequence of numerical values profiling certain biochemical properties along the whole genome strand. With a set of assays and a set of cell types we can then obtain a profile matrix as depicted as following:
Table 3: Profile matrix for cell and essay types.
A large profile matrix about various cell types can help us to understand the behaviors of those cells. However, it is currently rather expensive to conduct essays in large numbers. To alleviate such limitations, researchers have tried to build models based on available data, so that they can impute profiles for news cell types or essay types (ChromImpute, AVOCADO, PREDICTD, Encode Imputation Challenge,)
We see that table 3 has similar structure as table 1, thus we can model table 3 similarly as table 1. We notice here two main difference between the two tables: Firstly, table 3 is only sparsely defined as only limited number of experiments have been done. Secondly, an element in table 3 is a whole sequence (which may be potentially billions of numerical values.), whereas an element in table 1 contains a single value. It is obvious that the training algorithm for table 1 can handle the data sparsity problem without any change. In order to obtain representation for each cell and assay type, we only have to make sure that each row and each column contains some defined profile data.
In order to address the high dimensional data issue, we extend our neural network as depicted as following:
Figure 1: Neural network for cell- and assay-type representation learning.
The training algorithm runs roughly as following:
Randomly select a profile from table 3, say a profile P(A, b) for cell-type A and assay type b.
Segment the profile into sections of equal length N; and arrange the sections into a matrix PA,b that has L rows and N columns.
Lookup the representation RA for cell type A in present set of representations. If no such representation is found, initialize RA as a zero-matrix of dimension LxDc, where Dc is a preset constant.
Lookup the representation Rb for assay type b in present set of representations. If no such representation is found, initialize Rb as a zero-matrix of dimension LxDa, where Da is a preset constant.
Randomly select an integer k between 0 and L; Push the k -th row of RA and Rb into the input layer of the network; Run the optimization process, e.g. with ADAM optimizer, for one step with k-th of PA,b as the target label.
Pull back the state of the input layer into the k -th row of RA and Rb respectively.
Repeat the steps 5 and 6 for certain preset numbers.
Select an other profile and repeat step 1 to 7 till the training process converges to certain state with small errors.
Notice that the network depicted above consists mainly of dense feed-forward layers. Only the last layer is a transposed convolution layer. This layer significantly extends the output dimension and therefore speedup the training process for long profile sequences. Another technique to speedup the training, that is not directly visible in above diagram, is the batched learning: the training process updates the weight matrix and input layer states only after 30 to 40 samples have been feed into the algorithm.
Representation Learning versus Model Learning
Most deep learning frameworks are model learning in the sense that they train models to learn data. Compared to the amount of training data, the size of the trained models are relatively small. RL extends model training to learn vector representation for key entities, which subject to certain algebraic relationships. Those key entities can be, element of mathematical groups, 3D image transformation, biological cell and assay types. Since RL exploits modeling power of deep neural network in the same way as model learning does, methods and techniques developed for model learning (like data batching, normalization and regularization,) can be directly used by RL framework.
It is interesting to notice RL incidentally resembles the working mechanism in cellular organism as revealed in molecular biology. In the latter case, the basic machinery of a cell is the RNA ribosome, which is relatively simple and generic compared to the DNA sequences. The ribosome could be considered as a biological model, whereas the DNA sequences as representation (i.e. code) for plethora of organic phenotypes. The learning process of the two models are however different. Whereas the ribosome model learns by recombination and optimizing selection, RL learns by gradient back-propagation. The former seems to be more general, whereas the latter be more specific and, arguably, more efficient. Nevertheless, I do think that nature organism at certain level might host more efficient gradient directed learning procedure.
Conclusion
We have presented a representation learning method with deep neural network. The method basically swaps the data representation with states of neuron nodes during the training process. Intuitively, those learned representation can be considered as target data back-propagated to designated neuron nodes. With examples we have demonstrated that RL algorithm can effectively learn abstract data like algebraic entities, transformations, and can perform services like dimensionality reduction and data imputation.