Multidimensional Sorting with High Fidelity t-SNE
Multidimensional data often has a latent sequential structure that determines the basic shape of the data. Those sequential structure might correlate with ordered quantities like time, stages and size etc. Capturing and visualizing those sequential structure would be very helpful for exploratory data analysis.
A frequently used method for exploring high dimensional data is the principal component analysis (PCA). By projecting data to the main principal components, we can gain institutive information about the high level large scale shape of the data. More particularly, the projection to the first principal component offers a way to visualize the main back-bone linear structure.
A main short-coming of PCA is that it only structure from linear perspective. It fails for data whose latent sequential structure significantly deviate from linearity. For instance, let's consider the 2-dimensional data set depicted by the following map:

This dataset consists of multiple clusters each possesses, more or less, a sequential structure. The PCA wouldn't be able to aggerate these partial sequential structure to a global sequential structure.
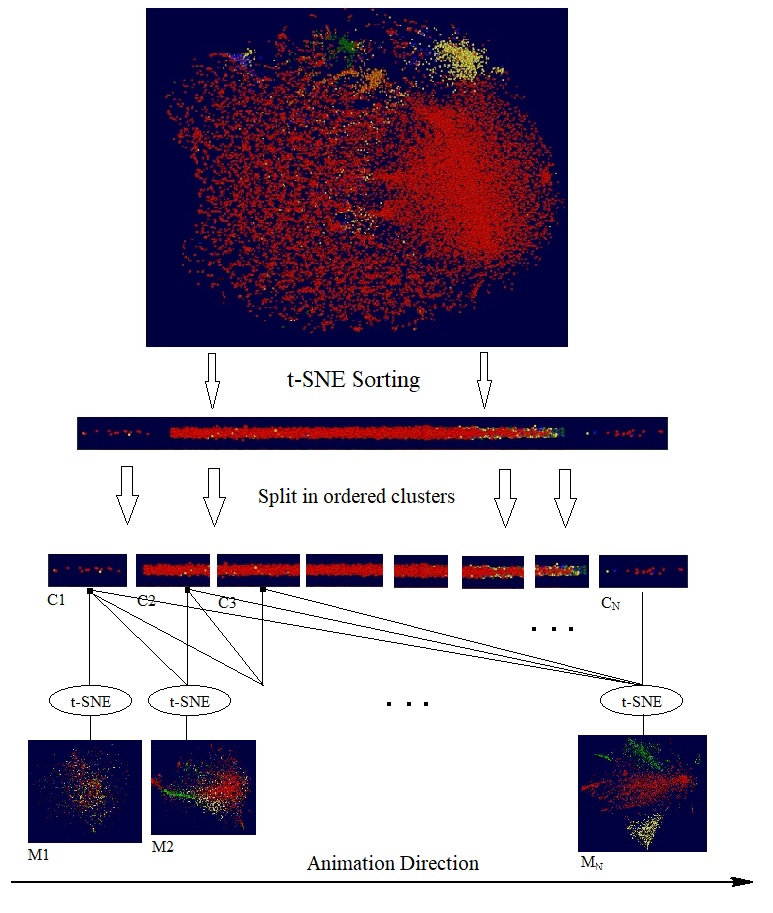
The t-SNE algorithm with the high fidelity modification offers a simple yet effective way to discover global sequential structure in high dimension data: we simplify apply t-SNE to reduce a given dataset to 1-dimensional space. The coordinates of data points in the 1-dimensional space then provide the keys to order data points. The following video clip illustrates how t-SNE works on the dataset mentioned above:
We notice in above video that t-SNE discovered, more or less, the sequential structure underlying the sub-clusters; and furthermore, those partial sequential structures are aligned/ordered in a coherent way.
Dual Sorting of Heatmaps
Heatmap is a popular method to visualize matrix of numerical data. Heatmap is often used together with dendrograms, which rearrange the rows and columns using certain hierarchic clustering algorithm. The following picture shows a typical heatmap together with two dendrograms:

Heatmap augmented with dendrograms (wikipedia)
Heatmap constructed with dendrograms offers intuitive visualization, but it has difficulty to scale up to large dataset. For large matrix like those from single cell RNA sequencing (scRNA seq) study, dendrogram become quite inefficient, and instable in the sense that small change in data might lead to large variation in the clustering structure.
The t-SNE based sorting algorithm described in this note provides a simple alternative to arrange rows and columns in a heatmap: we simply sort the rows and columns with t-SNE. As an example, I took a dataset from scRNA Seq analysis, it comprises the gene expression counts of about 11000 cells with respect to 28000 genes. The following pictures shows its t-SNE embedding and its heatmap:


- The perplexity of the t-SNE is set to about 2500. Smaller perplexity often leads to instability and random artifacts.
- The training process with t-SNE has ran for 5000 epochs, which is much higher than normal t-SNE training process. Training less epochs likely results in more random artifacts.
- In above example, t-SNE used the linear correlation as distance metric instead of the normally used Euclidean distance. Various experiments indicate that correlation seems to be more appropriate to measure similarity between genes. Because of this, we can not use PCA to pre-process data table to much lower dimensions to speed up the training, since PCA in general doesn't preserve correlation table.